

Practical Planning and Scheduling for Capital Project Delivery in the Pharmaceutical Industry
Francisco Cruz Moreno, PE
Abstract–Initiating a pharmaceutical capital project requires a deep understanding of the unique processes related to current good manufacturing practices, along with experience in the design, procurement, construction, commissioning, qualification, validation of facilities, utilities, and equipment. Additionally, planning should account for the regulatory affairs that approve bringing a product to market in this highly regulated industry. While most of the pharmaceutical capital projects are schedule-driven in a first-to-market business model, improper planning and delays constantly jeopardize the release of new products. This article describes practical techniques to plan and schedule new construction and renovations of existing buildings; typical sequences and durations used for commissioning, qualification, and validation activities; timing for engagement with different stakeholders; common pharmaceutical terminology employed during all phases of a project; and recommended approaches to leverage previously performed work to potentially gain time in the project schedule. Novice and seasoned practitioners alike will benefit from following these procedures. The author has successfully used these practices in manufacturing facilities for oral, parenteral, and topical administration products located in the Americas, Europe, and Asia. This article was first presented as PS-3597 at the 2021 AACE International Conference & Expo.
Introduction and Background
The global pharmaceutical industry is projected to grow during the upcoming years due, in part, to an aging and growing population, rising income levels, new diseases, and emerging medical conditions [1, p. 1]. While the pharmaceutical industry is one of the most regulated and capital-intensive on the planet, it is also more profitable than any other large public company [2, p. 841]. Since these companies operate in an increasingly competitive market, they have adopted creative business strategies to expedite product delivery. The current SARS-CoV-2 pandemic is just one example of how the pharmaceutical industry can quickly scale research, development, and manufacturing processes to be first to market with a product. The pandemic environment also supports the assertions as to why most pharmaceutical capital projects are schedule driven.
The term pharmaceutical as used in this article refers to the pharmaceutical, biotechnology, and nutraceutical sub-industries as they relate to the use of chemical and biological compounds, dietary supplements, and food additives. Furthermore, the term includes capital projects associated with oral, parenteral, dermal, and injection applications and research projects, including vivarium and process development.
Facilities and engineering teams work on a variety of capital projects to address the needs related to products and processes specific to this industry. These projects may include new production or support facilities, central utility plant upgrades, new cleanrooms or renovations, operational expansions, interior fit-outs, technology transfers, automation, and building and environmental management systems. Projects can be executed on one site, within one building or across multiple buildings; or on multiple sites, located in the same country or across the globe; and can be completed concurrently or sequentially.
These teams face common challenges as they begin to plan and execute projects. Common denominators include project complexity; massive coordination with multiple workstreams; stringent room environmental conditions; qualified workforce resources; changing equipment and software technologies; rigorous documentation, validation, and regulatory requirements; operational consolidation projects; and multiple company reorganizations. The fact that these capital projects may be executed in different countries adds to the complexity since the process requires qualifying designers, contractors, vendors, and suppliers to ensure that they are familiar with the industry’s quality, environment, and documentation requirements.
This article presents practical planning and scheduling techniques that address these challenges by focusing on the timing for engagement with different workstream leaders; typical sequences and durations used for commissioning, qualification, and validation (CQV) activities; and recommended approaches to leverage previously performed work to potentially gain time in the project schedule. The author uses pharmaceutical terminology familiar to all phases of a project. This document will not cover the business process, sanction of candidate products, procurement of architectural and engineering services, or supply chain management after products are approved by regulatory agencies as these are company-specific and tend to vary widely.
Phases of a Pharmaceutical Capital Project
Pharmaceutical projects require additional phases that are not common in other industries since they need to maintain the integrity, quality, and safety of their processes and products by ensuring that they meet current good manufacturing practice (CGMP) and government regulatory requirements. In addition to design, procurement, construction, and commissioning, these projects must go through qualification, validation, and regulatory phases. Project teams ensure that the asset meets not only the design intent and requirements but, most importantly, the manufacturing processes and the product.
The project scope definition and planning stages follow a phase-gate process where front-end loading (FEL) and a project definition rating index are the most widely used rating systems. Steering committee members evaluate projects at every gate and decide whether to approve them, place them on hold, ask for a reevaluation, or cancel them. Projects with weak scope definition fail to secure approval and funding to move to the next gate. To secure approval, deliverables must be organized, and the first two gates should be conducted with discipline and additional rigor.
To be successful, the project team should start a coordination effort and an integrated documentation process as soon as practically possible after project kick-off. Identifying the departments and leads that will be involved on the project should start as soon as the project is sanctioned; the goal is to create a core team that reports to a project sponsor and to a steering committee. This team will identify, collect, confirm, and document the project requirements. A typical team composition is shown in Table 1.
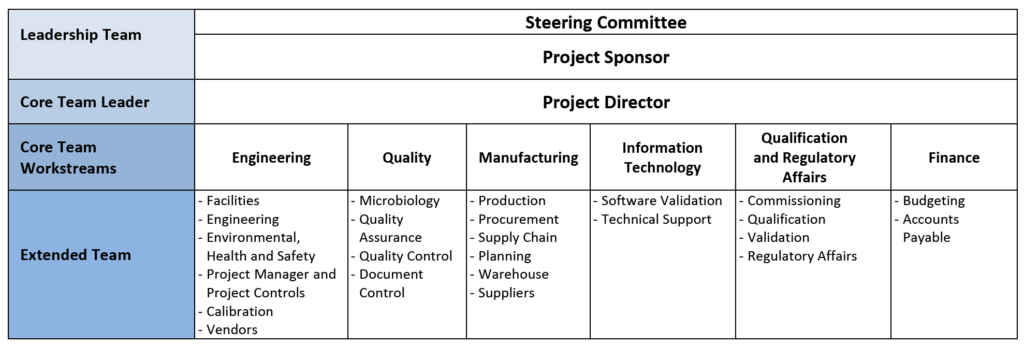
Table 1–Typical Project Team Composition
Owing to the regulatory requirements, the project manager, along with the CQV team, will start reviewing and updating the validation master plan (VMP). This is the document that defines and documents the elements of the site’s validation and qualification program. The contents of the VMP, or an equivalent document, are often neglected during the early planning stages and are not included in the project schedule. To mitigate this issue, the project team should review and amend the VMP as soon as there is an approval to any significant change to equipment, a section of the facility, processes, or testing methods that could affect the quality of the product.
Before the design phase begins, the planning and scheduling professional should review and understand the following items within the VMP as part of the project schedule planning and development:
- Validation philosophy
- Requirements for establishing validation (e.g., direct and indirect product impact, and critical and noncritical components)
- Site process and product description
- Approval levels required for documentation
- Inventory of products
- Manufacturing processes
- Systems to be validated (e.g., environmental management system)
- Planning and scheduling of the validation activities
Design
The design of a pharmaceutical facility addresses key features, including adequate space to facilitate the functions and efficiency of manufacturing, cleaning, and maintenance operations; sustain specific environmental conditions related to temperature, humidity, and differential pressure; prevent cross contamination between adjacent areas, corridors, and rooms; minimize exposure to hazardous material; and enable the proper flow of personnel, materials, and waste. During this phase, the project team starts collecting information, such as user requirement specifications (URS); functional requirement specifications (FRS); software requirement specifications (SRS); budget, product volumes, schedules, and key milestones; utility and safety requirements; and standard operating procedures (SOP).
A typical work breakdown structure includes disciplines related to architectural, civil, and structural; process and manufacturing equipment. This list includes utilities, piping, and instrumentation; automation, building automation systems, and environmental management systems (EMS); heating, ventilation, and air conditioning (HVAC), including air handling units (AHU) and fan coil units (FCU); mechanical services; fire alarm and fire protection systems; electrical, environmental, and health and safety systems; and information technology, cameras, and access control [3, p. Subpart C].
The permit strategy should be thoroughly assessed during the design phase since the site may require that additional environmental permits related to biological and process byproduct waste, discharge (or even treatment) be secured, potentially increasing the permitting duration. Meeting with the local building department to discuss the details of the project, understand the permitting requirements, and anticipate any additional demands can facilitate and expedite the permitting process. Additionally, some building departments have checklists to identify specific items that the department will review in the project drawings. The team can use these checklists during 90% design to provide feedback to the designer to anticipate potential design omissions. The author has used this approach to reduce the permitting schedule by one to two months.
The qualification team will perform a risk analysis and impact assessment (RA/IA) on product and equipment quality and process fitness. This will allow them to identify the systems that will be deemed direct or indirect impact, as well as critical and noncritical components. System boundaries are also identified as part of this assessment to assist the construction and CQV teams in organizing the documentation and in identifying responsibility assignments.
The outcome of the RA/IA typically classifies the documents into three categories: baseline documents, controlled documents, and validation documents. This key step should occur early during the design phase because documentation requirements may be different depending on the critical classification of the systems (e.g., good engineering practice guidelines may be sufficient to document noncritical components or indirect impact systems). The team should capture the development, execution, and approval of this documentation in the project schedule.
Besides the RA/IA, the team should perform at least three project risk analyses, specifically for Class 5, Class 3, and Class 1 schedules. Class 5 schedules contain limited information, and the team will most likely need to rely on limited historical data [4, p. 10]. While the project risk analysis focuses primarily on the facilities and equipment installation, the team should incorporate the risks identified for the product and processes, which are usually captured in a failure mode and effects analysis register.
The design can be executed in two stages (i.e., a combined schematic design/design development and construction documents) or in three stages (i.e., schematic design, design development, and construction documents), depending on the organization maturity, project size, procurement practices, and chosen project delivery method. Small projects that do not include process equipment usually fall under the two-stage design approach, whereas medium to mega projects fall under the three-stage design approach. The project team will hold design qualification (DQ) review meetings to ensure that the design captures all quality features, facility finishes, and flows; instrumentation performance; and functional criteria to move from one stage to the next. DQ also helps the engineering team make a decision regarding the equipment vendor selection.
The project team should explore the option of modularization and prefabrication, especially for sites where operations will run concurrent with new construction or renovations. During the mid-to-later stages of the design phase, the project team must decide what process and manufacturing equipment should undergo formal factory acceptance testing (FAT) and site acceptance testing (SAT). The team will also develop and approve FAT and SAT test protocols documentation; these activities should be included in the schedule and are considered pre-commissioning tasks.
The team will start planning the allocation of staff, resources, implementation strategies, and raw materials needed for the commissioning and qualification phases. The development of an integrated master schedule (IMS) is key to support the business plan and target the commercial date with a higher level of certainty. Figure 1 illustrates an IMS incorporating all major phases found in a typical capital project.
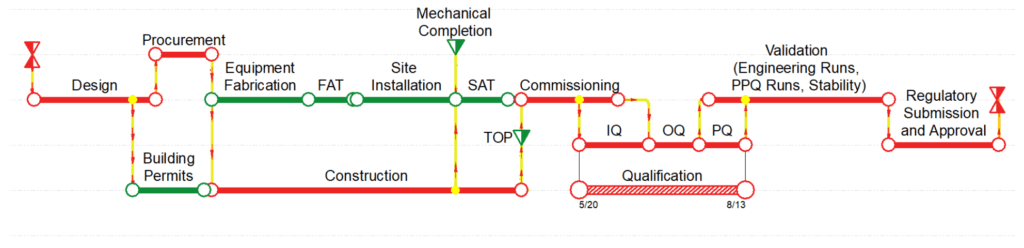
Figure 1–Common Schedule Sequence
Procurement
Besides procuring the services of a general contractor or a construction manager, the team must identify whether they will use in-house resources to perform and execute the commissioning and qualification (C&Q) protocols. If the project team decides to outsource the C&Q services, these consultants should be brought in early during the design process. For each equipment identified in the approved commissioning documentation, the commissioning team will perform an FAT at the manufacturer’s facility to ensure that the equipment operates as intended and that it complies with the URS, SOP, and design specifications (DS). A sample equipment and utilities list is shown in Table 2.
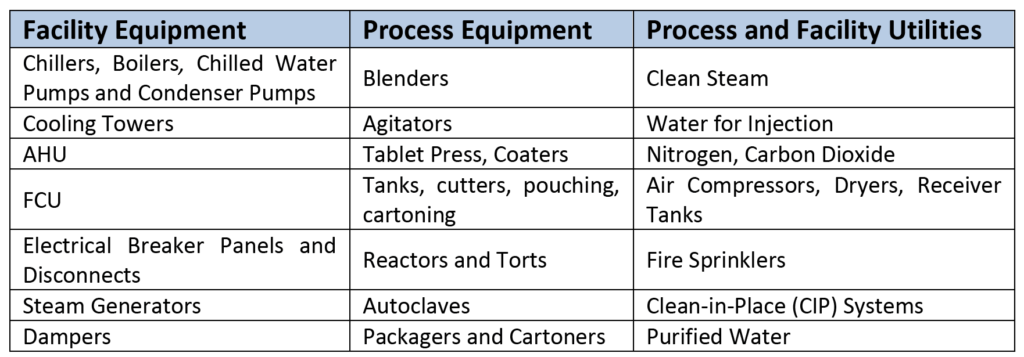
Table 2–Sample Equipment and Utilities List
Some of the functional testing will include drawing and wiring verification; instruments and calibration; utilities verification; safety, alarm, and security verification; and software quality assessment. Since process and manufacturing equipment are often deemed long-lead items, the team should include shipment and clearing of customs durations to reach the site. The project team will identify the commissioning team that will support FAT and SAT activities; this team could be composed of in-house personnel, consultants, or a combination of the two.
Construction
The project team will oversee the construction of each room and the installation of systems and devices, keeping quality as a paramount criterion for the completed area. Because there is minimal interaction with existing systems, production, and operations personnel, new construction usually has less coordination and constraints than renovation, additions, or remodeling.
For construction in existing manufacturing areas, the project team will coordinate with several departments, including facilities, manufacturing, quality, planning, and quality assurance to prevent and monitor the collection of airborne particles and dust; maintain the environmental conditions of adjacent rooms (i.e., temperature, humidity, differential pressure); and prevent vibrations to avoid recalibrating devices and equipment. The project team should install high-grade, opaque temporary partitions, seal space penetrations, and install differential pressure devices, such as magnehelics and photohelic gauges to monitor room pressurization. They should also install air scrubbers with high efficiency particulate air (HEPA) filters to maintain negative pressure and to avoid cross contamination between areas.
Additionally, construction personnel will receive training on avoiding triggering fire alarms and impairing the fire protection system following the site’s insurance carrier instructions. It is recommended that the project team perform a test and balance (TAB) verification of the existing HVAC system, with a goal of ensuring that environmental conditions required for manufacturing a product remain the same as before construction. A TAB must also be performed after construction completion. Implementing these preventive actions will ensure swift execution of the construction phase within the constraints of existing controlled areas.
The project team should also take advantage of site shutdowns that occur once or twice per year; these usually take place during the summer and winter seasons and can last between one to two weeks. There may be a need to request a shutdown for a longer period to install, commission, qualify, and validate a manufacturing area located in the core of a building, or, due to the nature of the process, the area may require extensive testing. In this case, in addition to getting approval from senior management, the project team should coordinate with the manufacturing team to build inventory and allocate space in the warehouse for additional finished goods.
The engineering team will coordinate with manufacturers and vendors to begin the SAT activities, which start after the confirmation of both mechanical completion and available site utilities. Note that some equipment manufacturers and vendors may have to travel from other countries to the construction site, so planning needs to occur well in advance. The commissioning team will start the equipment installation verification and functional testing to verify that the equipment operates as it did during the FAT.
It is recommended that the commissioning team witness the physical testing, perform visual examination, and document construction activities (e.g., loop testing, pressure testing, test and balance of HVAC, special welding) to expedite the commissioning process and receive certification. Additionally, the team could potentially leverage some of these testing results and certifications during the follow-on installation qualification (IQ) execution in the qualification phase.
Commissioning
Depending on the maturity of the organization, commissioning may be the last phase in which contractors and vendors provide support services. Refer to Figure 2. Mature companies will perform both qualification and validation tasks using in-house personnel. There should be milestones denoting handover activities as well as a period of knowledge transfer captured in the schedule.
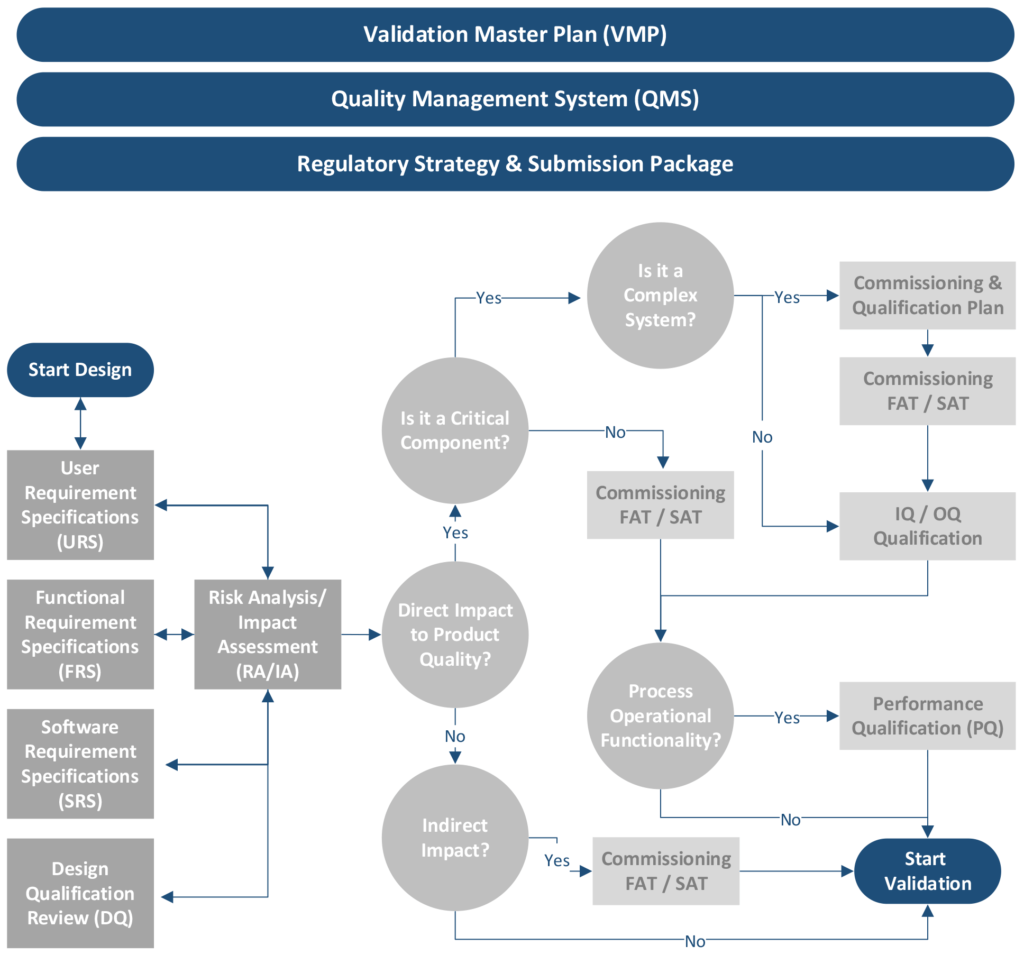
Figure 2–Flowchart of the Commissioning and Qualification Process
A successful commissioning phase provides documented evidence that the installed or modified equipment complies with the URS, FRS, approved design, and manufacturer recommendations. The commissioning activities include equipment and components testing, drawings and wiring verification, instrumentation and calibration, utilities verification, software assessment, security access, and personnel training. These may take from several days to two months depending on the complexity of the equipment and systems.
Although it may require more rigorous testing and inspection protocols, enhanced commissioning should be implemented whenever possible (i.e., usually for indirect impact systems) to avoid repeating activities during the qualification phase. Some commissioning activities may overlap with qualification activities (e.g., SAT and IQ).
Qualification
Qualification is the action of verifying, proving, and documenting, with a high degree of assurance, that the product, process, equipment, and ancillary systems are properly installed, work as specified, and lead to the anticipated results. Refer to Figure 2. The development of the qualification master plan (QMP) should start during the design phase and include a listing of protocols related to IQ, operational qualification (OQ), performance qualification (PQ), and resources that support the execution and documentation of the protocols. These protocols ensure that the equipment installation consistently meets the quality requirements of the finished product.
While qualification teams usually execute these protocols sequentially, the IQ and OQ protocols could be performed concurrently for systems already in operation in the case of equipment replacement, but the process remains the same; personnel may refer to it as the IOQ protocol. The deterministic duration of IQ, OQ, and PQ protocols depend on the complexity of the equipment, the qualification plan, and the experience of the team executing the protocols. The duration could take between twenty days to several months for each protocol.
The project team needs to account for material supply since the PQ could potentially use production material or a qualified substitute surrogate media for qualification testing. As the final step in the qualification phase, the qualification team will execute a PQ to test all equipment and components as a partial or completed manufacturing process; this is different from IQ and OQ where personnel test and verify each equipment and component one by one. The qualification team must complete the appropriate qualification of critical equipment and ancillary systems before starting validation activities.
Qualification of the HVAC system is required owing to potential impacts to product quality. Depending on the cleanroom classification, the qualification team will test several design features that include temperature and humidity thresholds and controls (T and RH); airborne particle control, room airflow direction, air changes (ACH), and differential pressure (DP); and HEPA filter integrity testing.
Table 3 shows key criteria that the qualification team needs to identify, plan for, and verify for each process equipment identified in the project. Generated reports provide the documented evidence and will be included as part of the regulatory submission package; these reports will be kept at the project site for potential regulatory audits.
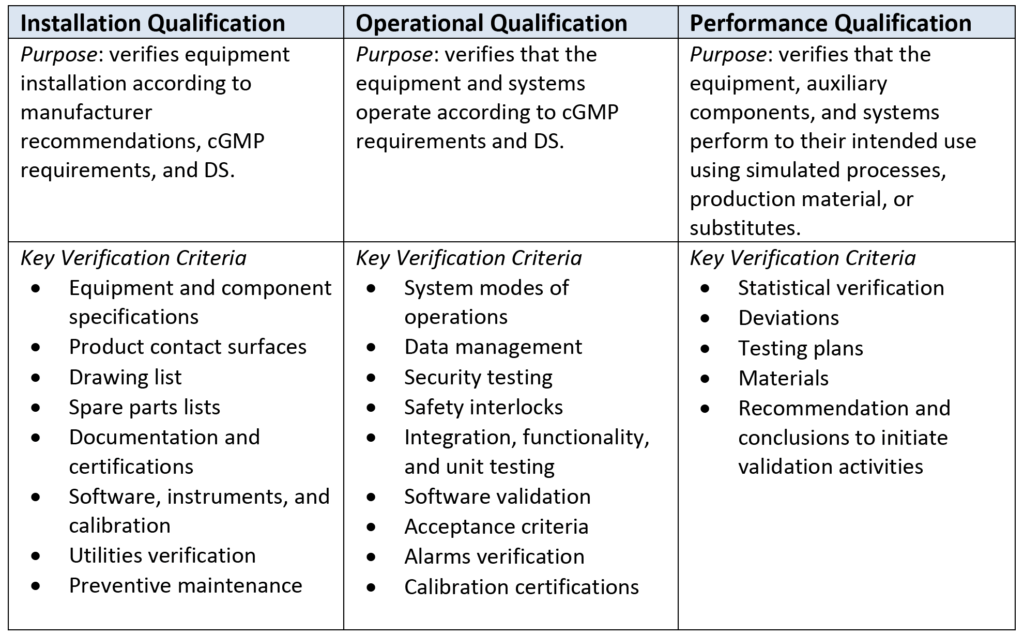
Table 3–Purpose and Key Criteria for Qualification Protocols
Since there may be a need to hire personnel to support a new area or manufacturing process, planning for the training of personnel in the processes, equipment, safety and handling of material and waste is key during this phase. These trained personnel will then execute the validation tasks during the next phase and will be responsible for the manufacturing process once the product receives approval from the regulatory authority.
Validation
Process validation refers to the collection and assessment of data to establish scientific evidence that a process can consistently deliver quality products. It is a regulatory requirement for any company aiming to obtain licensure of a pharmaceutical product. Process validation occurs from process design through commercial manufacturing and aims to detect and control variability in the manufacturing process. This is the final phase of a pharmaceutical project before submitting all documentation to the regulatory agency that will either approve or reject a product.
For new facilities, the team will use prospective process validation prior to manufacturing and selling a commercial product. For existing manufacturing operations and facility renovations, the team will use revalidation to maintain the current process validation while making changes to equipment, process, or product. There may be instances where the team needs to perform a retrospective process validation to assess the consistency of a process for products already in the market, but this approach may require several levels of approvals from senior management due to the increased level of effort required to execute it.
There is great emphasis on implementing well-understood control strategies and on demonstrating that the delivery team used a risk-based approach to identify critical process parameters. Some projects may include technology transfer efforts that require early planning and additional rigorous coordination between two or more sites, sometimes located in different countries. Transferring processes and products between countries also triggers additional regulatory requirements. Note: Due to the complexity of this subject, this article will not discuss technology transfer projects.
A main schedule risk driver that may impact the execution of validation tasks is the availability of skilled resources. Personnel assigned to carry out validation activities need to have the qualifications, training, and process experience related to cGMP and be familiar with the company’s SOP.
The team should also assess the cleaning validation procedure during the validation process. This validation is usually performed during the part of the process when risk of contamination or carryover of material that could compromise the product quality is the greatest. Cleaning validation is performed concurrently with product validation; however, the team may perform it prior to shorten the validation timeline. Existing validated cleaning procedures may not require additional validation if a new product belongs to the same classification and family type or if the product is a like-for-like replacement of existing equipment, pipes, and vessels (e.g., CIP systems).
The validation team is typically a cross-functional unit composed of key personnel from the quality control, quality assurance, manufacturing, product development, and regulatory departments. The validation team must plan to develop validation protocols using the site’s SOP, and the schedule should reflect at least the following activities: protocol development, review, and approval; protocol execution; protocol report generation, review, and approval; and validation documentation change control. Validation procedures and protocols must support the validation activities related to computer hardware and software, critical systems and equipment, process utilities, and facilities validation. The deterministic duration of the execution, report generation, and review and approval of a protocol typically ranges from five to 20 days.
There are additional key activities that occur as part of validation to confirm the manufacturing process. These activities are sequential in nature and there are few opportunities to perform them concurrently. They include engineering runs, process performance qualification (PPQ) runs, and stability periods.
Engineering runs are the initial runs, at the expected production scale, that simulate the formal CGMP manufacturing run using the actual qualified process equipment. The goals of the engineering runs are to confirm the successful scaling up of the new manufacturing process, complete the batch records [5, p. 210.3], vet the process control strategy, and train production personnel. Note that the validation team needs to plan the acquisition and disposition of material associated with these runs. The material could be either the same production material or what is referred to as water batch, which is material that may be composed of media or buffer. The quality control team oversees the engineering runs and performs full testing; the quality assurance team performs assessments and investigations on deviations that could affect product manufacturing. Depending on the risk appetite of the organization, engineering runs, along with their stability testing results, may be used as part of the submission package. This approach is considered mainly for manufacturing processes that have been operating for more than several years.
One of the most debated subjects within the validation phase is related to the appropriate number of PPQ batches. A decade ago, the gold standard was to run at least three PPQ batches to satisfy regulatory requirements. Now there are many different approaches in determining the number of PPQ batches after the FDA issued its new guidance in 2011 [6, p. 4] . The 2011 guidance is based on process experience and knowledge; statistics (e.g., expected coverage, process confidence, and capability); risk management; or a combination of these.
The validation team must identify the strategy and approach to run the PPQ batches before the end of the commissioning phase. Ideally, the number of PPQ batches may range between two and five depending on the product and robustness of the process. Note that the PPQ runs are considered commercial runs.
The last validation activities are related to collecting product stability testing data and developing reports to submit to regulatory agencies as shown in Figure 3. The validation and quality teams will evaluate the data from the PPQ runs to identify any deviations and degradation of the final product due to storage conditions (e.g., temperature, humidity, light); number and size of the batches; and closure systems [3]. The stability testing program must be defined in conjunction with the validation strategy and plan. The validation team may decide to collect stability testing data for the engineering runs to submit as part of the regulatory submission, potentially saving several months during the validation phase. The deterministic duration of the stability testing activities will vary depending on the product and process and will typically range between one, three, and six months.
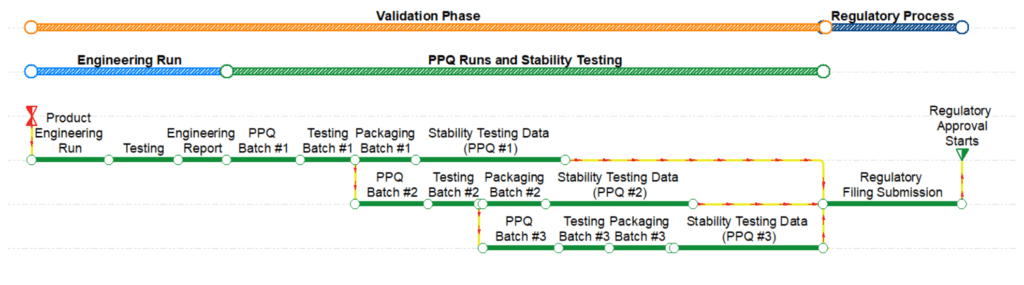
Figure 3–Schedule Showing the Validation and Regulatory Process Phases
Regulatory Process
One of the most important steps of a pharmaceutical project is to assess and ensure compliance of all laws and regulations pertaining to chemical and biological manufacturing products. A regulatory affairs (RA) department will define a regulatory strategy and prepare a regulatory submission package to request authorization to manufacture, promote, and sell the finished product. The regulatory strategy identifies whether a product will be registered with a domestic regulatory agency (e.g., United States Food and Drug Administration), or if it also will be registered internationally (e.g., European Medicines Agency in the European Union); the latter will increase the effort to comply with different regulatory requirements. These tasks may be outsourced to RA consultants; the core team must track them in the schedule.
The regulatory submission package not only includes the results from clinical studies and the stability testing data but also the commitments for post-registration and market surveillance to ensure patient protection. New products will most likely have a regulatory strategy defined and completed during Phase 1 or Phase 2 clinical trials. Existing products going through an increasing market demand, and that are approved for a facility expansion, have their regulatory strategy approved during the last stages of design of the facility. Regulatory approvals typically range from one month for existing products to twenty-four months for new products.
Change Control
A critical process within the pharmaceutical industry is related to the control of changes impacting the facilities, engineering, manufacturing, quality, research and development, and information technology departments. Additionally, this process needs to address whether changes are related to new or existing products, modification or decommissioning of critical equipment, new or revised drawings, or regulatory changes. The project team needs to initiate, document, approve, implement, track, and control these changes before starting the project. This is especially critical since systems and equipment can be impacted during the planning and execution of a capital project and these change control tasks should also be included in the project schedule. Once created, a change request may stay open for several months since it requires verification of satisfactory completion of tasks and subtasks to close it.
The qualification team, along with personnel from the quality assurance department, will use an RA/IA (see Figure 2) to initiate a change request and to determine which documentation, tests, verification levels, and approvals will be included. The project may need only one change with several subtasks, or it may need several change requests.
Conclusion
This article presented the unique phases found in a pharmaceutical capital project, identified the key team members along with their interaction throughout planning and execution, and displayed a flowchart to aid practitioners in comprehending the intricacies of the commissioning and qualification processes. A specific set of criteria were listed to verify and document the installation, operational, and performance qualification protocols based on the practical applications of a risk-based validation approach. Coordination and implementation of the change control process early in the project is key to meeting interim and finish milestones and avoid unintended delays.
Understanding the complexities of the processes, requirements, and regulations to which capital projects in the pharmaceutical industry need to comply increases the likelihood of achieving regulatory compliance and obtaining market approval. Early planning, extensive coordination, and solid documented evidence are essential to moving through the various project phases. Whereas there are traditional sequential approaches to plan and execute protocols and testing, this paper presents methodologies, including enhanced commissioning and concurrent IOQ, that could potentially shorten the project schedule while maintaining the robustness of the results.
Each design, procurement, and construction method, as well as discussed recommendations, is vital to accomplish a successful commissioning, qualification, and validation campaign of a full-scale CGMP run, especially on existing manufacturing sites. Scheduling activities using the sequence and durations presented during the CQV phases will give the project team a good starting point when planning, with the added value that they could anticipate and eliminate potential sources of risk, failure, and deviations.
- NAVADHI Market Research, “Global Pharmaceuticals Industry Analysis and Trends 2023,” 17 May 2019. [Online]. Available: https://www.navadhi.com/publications/global-pharmaceuticals-industry-analysis-and-trends-2023. [Accessed 19 December 2020].
- S. V. G. C. E. Ledley FD, “Profitability of Large Pharmaceutical Companies Compared With Other Large Public Companies,” JAMA, vol. 323, no. 9, pp. 834-843, 2020.
- FDA, “Electronic Code of Federal Regulations, Title 21, Part 211,” 19 January 2021. [Online]. Available: https://tinyurl.com/y3bwkm66. [Accessed 20 January 2021].
- AACE International, Recommended Practice No. 27R-03, Schedule Classification Systems, Morgantown, WV: AACE International, Latest revision.
- FDA, “Electronic Code of Federal Regulations, Title 21, Part 210,” 19 January 2021. [Online]. Available: https://tinyurl.com/y6rjhxdz. [Accessed 20 January 2021].
- United States Food and Drug Administration, “Guidance for Industry – Process Validation: General Principles and Practices, Revision 1,” FDA, 2011.
ABOUT THE AUTHOR
Francisco Cruz Moreno, PE, is with PMA Consultants. He can be contacted by email at: fcruz@pmaconsultants.com
Rate this post
Click on a star to rate it!
Average rating 4.7 / 5. Vote count: 15
No votes so far! Be the first to rate this post.
Hello,
I trust this message finds you in good health. I am reaching out to you on behalf of a consortium of esteemed Qatar investors with a keen interest in exploring promising investment opportunities. With a substantial capital allocation of over $250 million, they are eager to discuss and potentially collaborate on ventures that promise favorable returns on investment (ROI).
Our investors are open to diversifying their portfolio across various sectors, including but not limited to Real Estate, Agriculture, Food/Beverage Production, and the development of Shopping Malls. We are also receptive to exploring opportunities in other sectors that exhibit potential for growth and profitability.
In light of your expertise and track record, we believe that your insights and industry knowledge could be invaluable in identifying and executing sound investment strategies. We are prepared to offer highly competitive terms and a favorable percentage of the returns generated.
I kindly request the opportunity to discuss this potential partnership further. Please let me know your availability for a meeting, either in person or through a virtual platform, at your earliest convenience. We are flexible and willing to accommodate your schedule.
Your contributions and expertise will be pivotal in shaping the success of this venture, and we look forward to the possibility of working closely with you.
Thank you for considering this proposition, and I eagerly await your response.
Best Regards,
Paul Zarkas.